 Materiał genetyczny człowieka
znajduje się głównie w jądrze komórkowym, natomiast nośnikiem informacji
genetycznej jest kwas deoksyrybonukleinowy (DNA). Cała informacja genetyczna
człowieka (genom) zawiera się w 46 "jednostkach organizacyjnych",
czyli liniowych cząsteczkach DNA, które w trakcie podziałów tworzą chromosomy.
W prawidłowym organizmie każda komórka posiada taki sam zestaw chromosomów
(wyjątkiem są tylko niektóre wyspecjalizowane komórki). W normalnej komórce
somatycznej człowieka znajdują się 23 pary chromosomów homologicznych, w tym 22
pary chromosomów somatycznych (pary chromosomów od 1 do 22) i jedna para
chromosomów płciowych ( XX lub XY). Każda para chromosomów homologicznych
zawiera jeden chromosom odziedziczony od matki i jeden od ojca. Oznacza to, że
zdrowy człowiek posiada 46 chromosomów, czyli zestaw chromosomów
charakterystyczny dla danego gatunku (organizmu), zwany kariotypem [Srebniak,
Tomaszewska, 2008: 13-21].
Materiał genetyczny człowieka
znajduje się głównie w jądrze komórkowym, natomiast nośnikiem informacji
genetycznej jest kwas deoksyrybonukleinowy (DNA). Cała informacja genetyczna
człowieka (genom) zawiera się w 46 "jednostkach organizacyjnych",
czyli liniowych cząsteczkach DNA, które w trakcie podziałów tworzą chromosomy.
W prawidłowym organizmie każda komórka posiada taki sam zestaw chromosomów
(wyjątkiem są tylko niektóre wyspecjalizowane komórki). W normalnej komórce
somatycznej człowieka znajdują się 23 pary chromosomów homologicznych, w tym 22
pary chromosomów somatycznych (pary chromosomów od 1 do 22) i jedna para
chromosomów płciowych ( XX lub XY). Każda para chromosomów homologicznych
zawiera jeden chromosom odziedziczony od matki i jeden od ojca. Oznacza to, że
zdrowy człowiek posiada 46 chromosomów, czyli zestaw chromosomów
charakterystyczny dla danego gatunku (organizmu), zwany kariotypem [Srebniak,
Tomaszewska, 2008: 13-21].
Jeżeli
jednak występują dziedziczne lub niedziedziczne różnice pomiędzy komórkami
danego organizmu (z. wewnątrzosobnicza), osobnikami należącymi do tej samej
populacji (z. osobnicza) lub pomiędzy populacjami (z. grupowa) - występuje tzw.
zmienność. Wyróżnia się m.in. zmienność rekombinacyjną, fluktuacyjną (wzrost,
masa ciała, IQ, pigmentacja włosów i skóry), alternatywną (układ grupowy Rh) oraz
mutacyjną, która dzieli się na euploidie (autopoliploidie i allopoloploidie),
mutacje genowe i chromosomowe (strukturalne i liczbowe) [Drewa, Ferenc,
Bratkowska, Jakubczyk, 2003: 279]. Mutacja to każda zmiana w strukturze lub ilości DNA, może polegać na
zmianie jednej lub kilku zasad, na ubytku lub wstawieniu kilku zasad, na
zmianie liczby chromosomów lub na przegrupowaniu fragmentu chromosomu
[Zbytniewski, Dylewska, 2003: 532]. Mutacje (aberracje) chromosomowe mogą
powstawać w komórkach somatycznych lub gametach oraz mogą być dziedziczone lub
powstawać de novo [Drewa,
Ferenc, Bratkowska, Jakubczyk, 2003: 288]. Są to zaburzenia
struktury lub liczby chromosomów i z tego względu dzieli się je na:
·
aberracje liczbowe (mogą dotyczyć
pojedynczego chromosomu np. trisomia)
·
aberracje strukturalne (obejmują
różne zmiany w strukturze danego chromosomu)
Aberracje strukturalne obejmują zmiany i
zaburzenia prawidłowej struktury chromosomów, a są one wynikiem pęknięć i/lub
przeniesienia fragmentów chromosomów oraz połączenia ich w nowe konfiguracje. W
sytuacji, gdy nić DNA chromosomu pęka, powstają "lepkie" końce, które
mogą połączyć się z innym fragmentem nici DNA. Takie nieprawidłowe połączenie "lepkich"
końców nici DNA prowadzi do powstawania chromosomowych aberracji
strukturalnych, w których wyróżnia się:
·
translokacje
·
delecje
·
duplikacje
·
inwersje
·
izochromy
·
chromosomy pierścieniowe
(koliste) [Srebniak, Tomaszewska, 2008:
23-25].
Zespół
Ringa (inne nazwy: pol. zespół
pierścieniowego chromosomu 18, ang. ring
chromosome 18 syndrome, r(18) syndrome), jest
to zatem choroba genetyczna spowodowana chromosomową mutacją strukturalną, w
wyniku której powstaje chromosom pierścieniowy. Chromosom pierścieniowy
(kolisty) powstaje natomiast w wyniku pęknięcia, a następnie połączenia końców
chromosomu. Powstają one najczęściej z chromosomów 4, 13, 18 oraz chromosomu X.
Powodują występowanie licznych zaburzeń rozwojowych [Drewa, Ferenc,
Bratkowska, Jakubczyk, 2003: 288-292]. Owe pęknięcia występują jednocześnie na
ramieniu krótkim (18p) i długim (18q) chromosomu, a
pozostałe odsłonięte "lepkie" końce chromosomu zostają ze sobą
połączone, tworząc pierścień [Srebniak,
Tomaszewska, 2008: 27]. Rezultatem jest przegrupowanie materiału genetycznego,
a konsekwencje kliniczne zależą od tego jaki materiał genetyczny uległ
usunięciu przed połączeniem ramion w "ring".
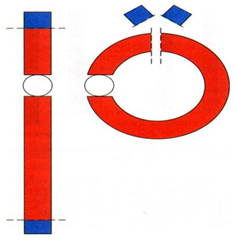
Na podstawie ryc. 15.15. Schemat powstawania chromosomu
kolistego [z:] Drewa G., Ferenc T., Bratkowska W., Jakubczyk M., 2003, Zmienność
i mutacje [w:] Podstawy genetyki dla
studentów, pod red. Drewa G., Ferenc T., Elsevier Urban & Partner,
Wrocław, s. 292
W
1962 roku
Wang (et al.) przedstawił swoją pierwszą obserwację na temat
zespołu pierścieniowego chromosomu 18, która zawierała cechy kliniczne r (18)
takie jak:
1. Wzrost i rozwój
1. Wzrost i rozwój
·
Upośledzenie umysłowe
·
Hipotonia
·
Niskorosłość
2. Głowa/ mózg
·
Mikrocefalia (małogłowie)
·
Dolichocefalia (długogłowie)
·
Holoprosencefalia
·
Cebocefalia
·
Arhinencefalia
3. Środkowa część
twarzy
·
Spłaszczona
4. Uszy
·
Asymetryczne
·
Nisko
umieszczone
·
Zarośnięcie
zewnętrznych kanałów słuchowych
·
Wystająca spirala
5. Oczy
·
Głęboko
osadzone
·
Hiperteloryzm
(zwiększona odległość między oczami)
·
Skośne
ustawienie szpar powiekowych
·
Zmarszczki
nakątne
·
Oczopląs
·
Zez
·
Ptoza -
opadnięcie powieki
·
Colomoba -
szczelina, rozszczep anatomicznych struktur oka
6. Nos
·
Szeroka
podstawa
·
Obniżony grzbiet
nosa
·
Szeroki czubek
7. Usta
·
Mikrognacja - nieprawidłowo mała żuchwa
·
Kąciki ust skierowane ku dołowi (tzw.karpie
usta)
·
Cienkie wargi górne i dolne
·
Wysokie / wąskie podniebienie
·
Próchnica
8. Szyja
·
Krótka
9. Skrzynia
·
Klatka piersiowa lejkowata
·
Szeroko rozstawione sutki
10. Wrodzona
wada serca
·
PDA
·
PS
·
VSD
·
AS
11. Brzuch
·
Przepuklina pępowinowa
·
Przepuklina pachwinowa
12. Układ
moczowo-płciowy
·
Genitalia męskie
a) Wnętrostwo - niewłaściwe umieszczenie jąder np. w jamie brzusznej
b) Hipoplastyczna moszna (niedorozwój moszny)
b) Hipoplastyczna moszna (niedorozwój moszny)
c) Mikropenis
·
Genitalia kobiece
a) Niedorozwój mniejszych warg sromowych
13. Zaburzenia mięśniowo-szkieletowe
·
Kręgosłup: skolioza
·
Dłonie
a) Klinodaktylia
(boczne lub przyśrodkowe skrzywienie palca)
b)
Kamptodaktylia (przykurcz zgięciowy jednego lub wielu palców)
c) Krótkie palce
(brachydaktylia)
d) Długie,
zwężające się palce
e) Bliższe
położenie kciuków
f) Pojedyncza
bruzda zgięciowa dłoni
·
Nogi/stopy
a)
Szpotawość
b) Szerokie/długie
palce
c) Koślawe
kolana - Genu valgum
14. Inne funkcje
·
Niedoczynność
tarczycy
·
Niedoczynność
przytarczyc
·
Niedobór
hormonu wzrostu
15. Nietypowe funkcje
·
Zespół Woude
van der
·
Insulinoniezależna
cukrzyca
·
Agammaglobulinemia
Większość
pacjentów wykazuje cechy zespołu del (18q):
a. Upośledzenie umysłowe
b. Hipotonia
c. Małogłowie
d. Niskorosłość
e. Środkowa część twarzy spłaszczona
f. Usta w kształcie karpia
g. Zwężenie
kanału słuchowego
Niektórzy pacjenci mają cechy del (18p), które są w wysokim stopniu zróżnicowane:
a. Upośledzenie
umysłowe
b. Opóźnienie mowy
c. Hipotonia
d. Niskorosłość
e. Wady twarzy w tym
holoprosencefalia (niedokonany podział przodomózgowia)
f. Opadanie powiek
g. Mała żuchwa
h. Próchnica
i. Krótka szyja
j. Niedobór IgA [Chen, 2006: 831-834].
Zespół
Ringa charakteryzuje się fenotypem bardzo zmiennym, w zależności od stopnia
usunięcia fragmentów 18p i 18q. Większość pacjentów wykazuje cechy
kliniczne zespołu delecji 18q, a
niewielka liczba pacjentów wyposażona jest w cechy kliniczne zespołu delecji 18p. Zazwyczaj jednak obserwuje się
połączenie cech klinicznych zespołu delecji 18p i 18q. Chromosomy
pierścieniowe występują niezwykle rzadko z częstością 1 na 50.000 żywych
urodzeń. W piśmiennictwie opisano dotychczas ok. 70 przypadków zespołu
pierścieniowego chromosomu 18, które w większości powstały de novo, choć zostało opisanych także kilka przypadków rodzinnych [Pereza, Buretić-Tomljanović,
Vraneković, Ostojić, Kapović, 2010: 208-213].
Bibliografia:
1/ Chen H., 2006, R (18) Syndrome
[w:] Atlas of Genetic Diagnosis and Counselling, Humana Press, Totowa,
New Jersey, s. 831-834
2/
Drewa G., Ferenc T., Bratkowska W., Jakubczyk M., 2003, Zmienność
i mutacje [w:] Podstawy genetyki dla
studentów, pod red. Drewa G., Ferenc T., Elsevier Urban & Partner,
Wrocław, s. 279-292
3/ Pereza
N., Buretić-Tomljanović A., Vraneković
J., Ostojić S., Kapović M., 2010, Ring
chromosome 18 syndrome, Medicina Fluminensis, Vol. 46 No. 2, 2010 [online:]
http://hrcak.srce.hr/file/81212, s. 208-213
4/ Srebniak M., Tomaszewska A., 2008, Badania cytogenetyczne w praktyce klinicznej,
Wydawnictwo Lekarskie PZWL, Warszawa, s. 13-27
5/ Zbytniewski Z., Dylewska K., 2003, Genetyka a ewolucja [w:] Podstawy
genetyki dla studentów, pod red. Drewa G., Ferenc T., Elsevier Urban &
Partner, Wrocław, s. 532
Komentarze
Prześlij komentarz